
M WANG Lab

Human genetics

Traditionally, vaccine effectiveness (VE) is estimated only after the population has been vaccinated and a portion of subjects have been infected. We establish the VE prediction method by modelling the relationship between VE and genetic distance (GD) of circulating viruses against vaccine strain, which allows immediate in silico prediction of VE before a new genetic variant enters a population.
The method enables timely evaluation of existing cohort vaccine protection to facilitate public health policy making and vaccine strain selection.
The initial statistical framework of VE-GD relationship is developed for influenza vaccine (Cao et al, 2021, Vaccine). One major issue in this study is determining the key mutations influencing VE, and aggregation model of the multivalent vaccine. Many of the challenge in modelling influenza vaccine haven’t emerged for the COVID-19 vaccines.
Cao L, Lou J, Zhao S, Chan RW, Chan M, Wu WK, Chong MK, Zee BC, Yeoh EK, Wong SY, Chan PK, Wang MH*. In silico prediction of influenza vaccine effectiveness by sequence analysis. Vaccine. 2021 Feb 12;39(7):1030-4.
Cao L, Zhao S, Lou J, Zheng H, Chan RW, Chong MK, Chen Z, Chan PK, Zee BC, Wang MH*. Differential Influence of Age on the Relationship between Genetic Mismatch and A (H1N1) pdm09 Vaccine Effectiveness. Viruses. 2021 Apr;13(4):619
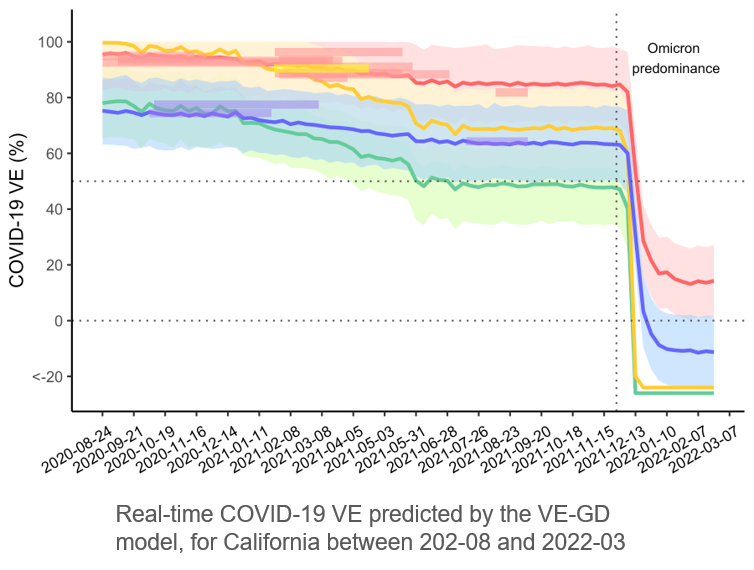
Based on the primary model we established for the influenza vaccines, we expand the framework for COVID-19 VE prediction. Unlike the influenza vaccines that are mostly manufactured by egg-based manufacture platforms, the COVID-19 vaccines adopt diversified technologies, which is the main variation to control in the modelling process. Meanwhile, analysis of SARS-CoV-2 genome benefits from the abundancy of datasets and the number of VE studies. The prediction accuracy by the proposed VE-GD model on independent validation dataset reached 95%.
Cao L, Lou J, Chan SY, Zheng H, Liu C, Zhao S, Li Q, Mok KP, Chan SY, Chan RWY, Chong MKC, Wu WKK, Chen Z, Wong ELY, Chan PKY, Zee BCY, Yeoh EK, Wang MH* (2022) Rapid evaluation of COVID-19 vaccine effectiveness against symptomatic infection with SARS-CoV-2 variants by analysis of genetic distance. Nature Medicine.https://rdcu.be/cPNdp
While the human genome collected in a population exhibits a stable landscape of mutation distributions, virus genome sampled at different time points shows dynamic mutation profiles due to fast virus evolution.
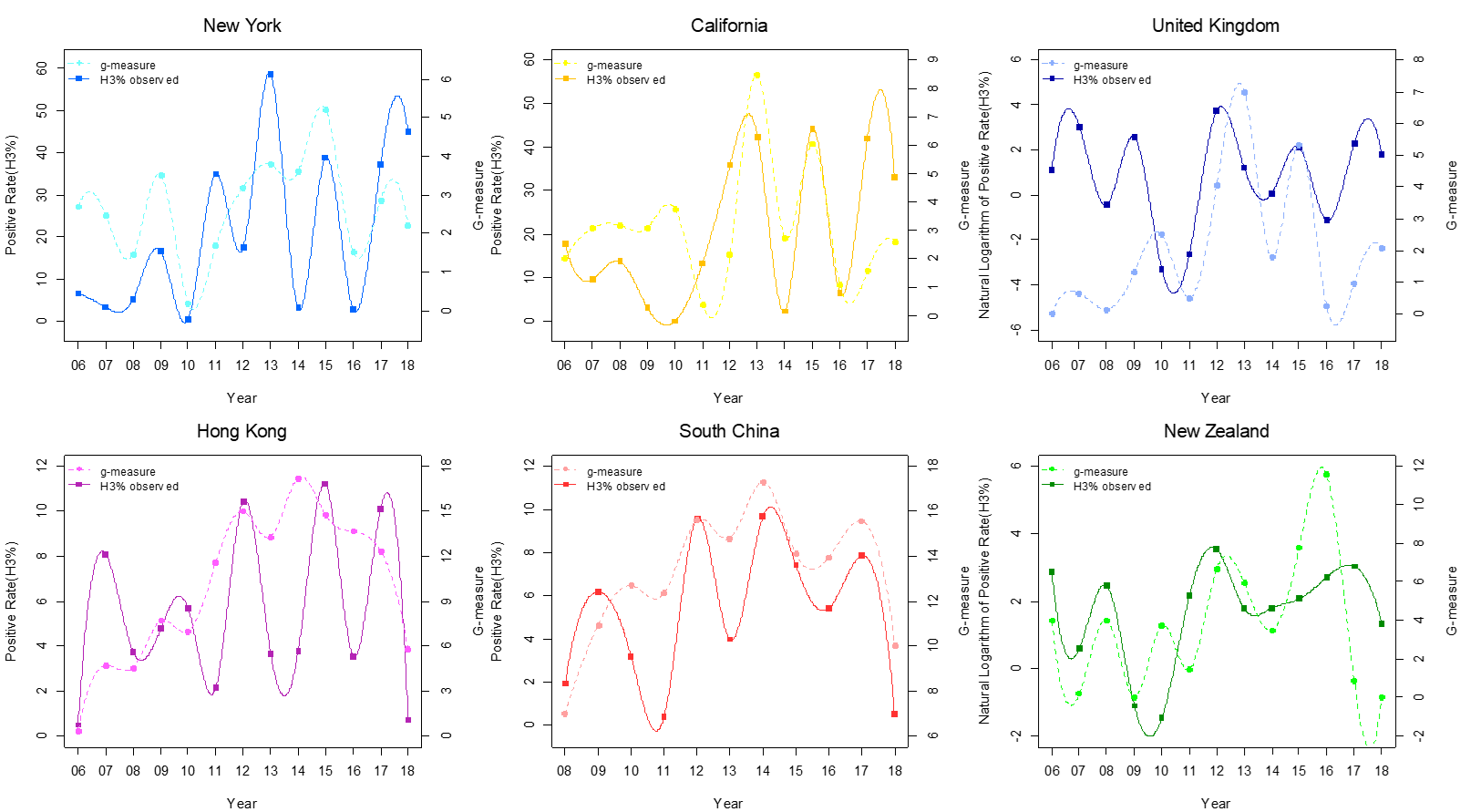
Clearly, summarizing the virus mutation activities through time will provide useful information about epidemics and virus evolution. Surprisingly, such a measure wasn’t unavailable in 2010s. Therefore, we introduced the g-measure (Wang Lou et al 2021, Journal of Infection), which is simply calculated as the sum of mutation prevalence of the virus in samples collected in a given time interval t.
gt = mt · pt
where m is an indicator variable (J-vector) of the key mutations over virus genome at time t, and p is the mutation prevalence in population at time t (J-vector). J is the genome size. Therefore, g summarizes overall mutation activities level in virus genome in a certain population within a given time period. The g-measure is shown to provide a good account of the population epidemic trend (see Figure in the right).
Primary method:
Wang MH, Lou J, Cao L, Zhao S, Chan RW, Chan PK, Chan MC, Chong MK, Wu WK, Wei Y, Zhang H, Zee BC, Yeoh EK (2021) Characterization of key amino acid substitutions and dynamics of the influenza virus H3N2 hemagglutinin. Journal of Infection. 2021 Oct
2018 US Provisional Patent No. 62/687,645, 18/MED/829, 2019 PCT/CN2019/091652 Measurement and Prediction on Influenza Virus Genetic Mutation Patterns. Wang MH, Zee BCY, Chong KC, Lou J.
Applications of the g-measure:
Lou J, Zhao S, Cao L, Chong MK, Chan RW, Chan PK, Zee BC, Yeoh EK, Wang MH *. Predicting the dominant influenza A serotype by quantifying mutation activities. International Journal of Infectious Diseases. 2020 Aug 22.
Lou J, Zheng H, Zhao S, Cao L, Wong ELY, Chen Z, Chan RWY, Chong MKC, Zee BCY, Chan PKS, Yoeh EK, Wang MH* (2022) Quantifying the effect of government interventions and virus mutations on transmission advantage during COVID-19 pandemic. Journal of Infection and Public Health.
We found that 80% of these EMs for influenza A(H3N2) HA were located in the antigenic sites, and the rest were located in the immuno-subdominant region HA2. This suggests that the statistically determined mutation sets might provide useful molecular candidates. Indeed, the EM sites offer the “features” that are predictive to influenza vaccine effectiveness, and enabled the VE prediction framework for influenza (Cao et al 2021, Vaccine)
As infectious disease pathogens evolves through time to escape from population adaptive immune responses, an once "effective" mutation that gives the pathogen selective advantage in the population will not be effective forever. Therefore, we recognize the necessity to propose the quantity"Effective mutation period(EMP)”, which captures for how long the key mutations would demonstrate advantage in the host population against its immune recognition. The EMP is jointly influenced by transmission dynamics, population density, immunity profiles in the population, and other epidemiological characteristics. The g-measure is dependent on the EM and EMP, thus it can capture the rises and falls of epidemic cycles over many seasons.
Prediction of influenza vaccine strains for the next epidemic season
A new method to predict influenza virus evolution and select vaccine strains for the influenza virus.
2018 US Provisional Patent No. 62/687,645, 18/MED/829, 2019 PCT/CN2019/091652 Measurement and Prediction on Influenza Virus Genetic Mutation Patterns.Wang MH, Zee BCY, Chong KC, Lou J.
Estimation for evolutionary rate
We develop a new probability framework to estimate virus substitution rate by genomic region, fast and effective, without building phylogeny.
More to be introduced…
A new method to perform codon optimization or deoptimization preserving viral structure information, with applications in improving efficiency of virus replication and antigenicity for vaccine production.
2021 US Provisional No: 63/283,91: Codon de-optimization or optimization using genetic architecture, Wang MH, Zheng H, Zee BCY
More to be introduced…